Multiwfn的轨道定域化功能的使用以及与NBO、AdNDP分析的对比(6)
时间:2019-04-11 14:55 来源:互联网 作者:阿刁 点击:次
默认情况下,NBO只能自动搜索单、双中心轨道,实际上,如果用pop=nboread,然后在输入文件末尾空一行写上$NBO 3cbond $END,则程序还会自动搜索三中心轨道。然而,这么做了之后结果更诡异,轨道分布根本不满足体系点群对称性,对体系电子结构的描述完全错误。NBO会搜索出的两个占据数为1.498的三中心轨道,图形如下
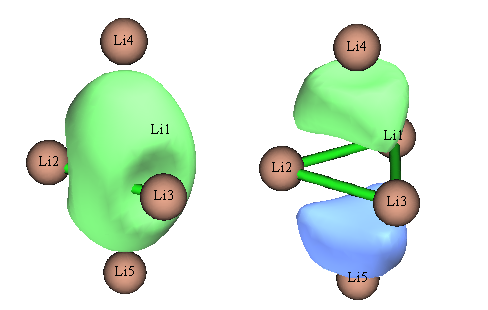
除了这两个外,NBO还搜出Li1有两个占据数分别为0.551和0.370的LP*轨道。 NBO6相对于NBO3.1在默认的轨道搜索规则上做了很大修改,但搞出来的NBO轨道也是完全没法用来讨论Li5+的成键本质。虽然NBO6已经允许在$CHOOSE中指定>3中心的定向搜索,但是却不能自动搜索>3中心轨道。所以对于存在>3中心轨道的情况,NBO几乎是没有丝毫用处的,用了只会被误导。若是信誓旦旦地拿这些NBO轨道去说事,明显是坑爹。然而很多初学者缺乏最基本的理论常识,程序输出什么结果就拿什么结果讨论,胡乱解释一番。盲目用NBO危害实在太大。 对此体系如果尝试用NBO3.1做NLMO分析,程序会报错,这是NBO3.1的bug所致。笔者也用NBO6做了NLMO分析,Li的2s轨道构成的NLMO轨道如下
实际上,这正是Li5+能量最高的两个占据MO的图形,NLMO等于完全没起到定域化效果!
此例我们考察OsO4的轨道,也是要得到轨道能量。输入文件如下 有兴趣者可以直接载入OsO4.fch,会看到从其中的MO图形上根本讨论不了此体系的成键特征。启动Multiwfn,依次输入
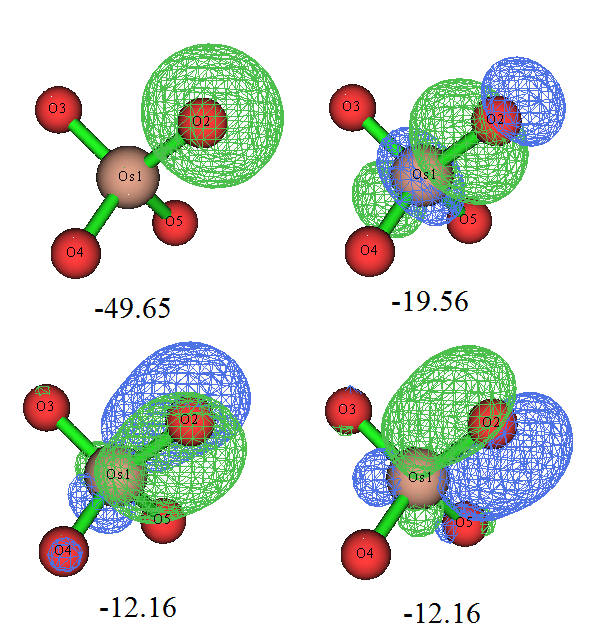
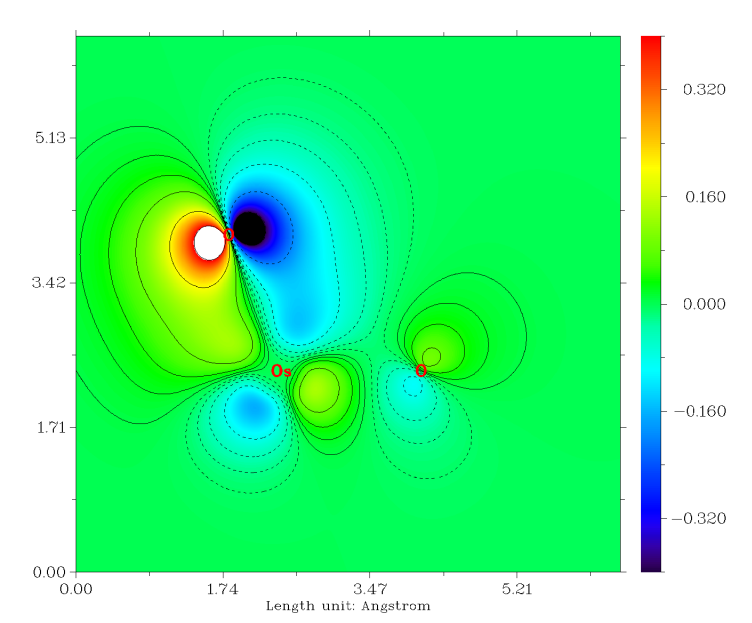
轨道图形和我们期望的很一致,而且轨道能量也很符合化学直觉,即sigma轨道能量低于pi轨道。那个孤对电子轨道几乎是球形的,如果用Multiwfn对它用Mulliken方法做一下轨道成分分析,也会看到它几乎完全由S基函数构成,很容易理解这就是氧的2s轨道。本身由于2s轨道的能级显著低于用于成键的2p,所以这个轨道的能量自然而然比sigma、pi轨道低得多。 虽然Os和每个O之间有3条成键LMO,但是也不代表它们的键级就是3.0,这只能认为是键级的上限。计算Mayer键级的话,发现Os和O之间的键级是1.720。如果同上一节做键级分解的话,会发现Os-O的sigma LMO对键级的贡献是0.866,而每个pi LMO的贡献仅有0.425,这可以认为这些pi轨道并不是在Os和O之间很充分共享的。我们可以在看轨道的时候把等值面数值加大,会发现在较大数值的时候,等值面几乎都出现在氧上。如果我们用Multiwfn的主功能4对pi轨道作个填色+等值线的截面图(相关操作参考手册4.4节绘制平面图的例子),可以将其分布特征看得更清楚,如下所示
|