启动Multiwfn,依次输入
Li5+.fch
19
-4 //Multiwfn将从此文件中读取Fock/KS矩阵
Li5+.47 //含有Fock/KS矩阵的文本文件
1 //对占据轨道做定域化
定域化收敛后,输出了LMO的能量信息,如下所示
1 Energy: -2.1347452 a.u. -58.0894 eV Type: A+B Occ: 2.0
2 Energy: -2.1347452 a.u. -58.0894 eV Type: A+B Occ: 2.0
3 Energy: -2.1347452 a.u. -58.0894 eV Type: A+B Occ: 2.0
4 Energy: -2.1213723 a.u. -57.7255 eV Type: A+B Occ: 2.0
5 Energy: -2.1213710 a.u. -57.7254 eV Type: A+B Occ: 2.0
6 Energy: -0.3037220 a.u. -8.2647 eV Type: A+B Occ: 2.0
7 Energy: -0.3037206 a.u. -8.2647 eV Type: A+B Occ: 2.0
当前内存中记录的占据轨道的能量,以及导出的new.fch里的占据轨道能量都是这些新算出来的能量。由于这里没对空轨道做定域化,所以它们的能量没被输出,内存里和new.fch里的空轨道的能量还是MO的。
从上面列的7个占据的LMO的能量来看,轨道分为三种,简并度分别为3、2、2。我们用主功能0观看轨道图形,每种简并的轨道只在下图绘制其中一个,图上标的是eV为单位的能量
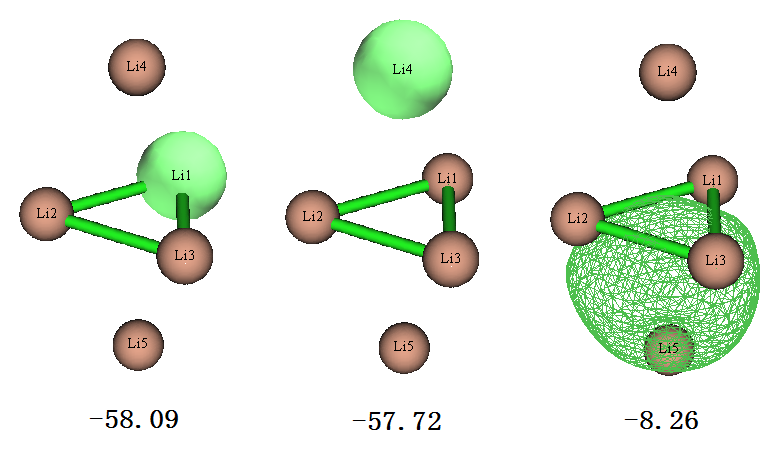
上图第一个轨道是中间位置的Li的单中心轨道,由于其能量很低,很容判断出这肯定是Li的1s轨道。上图第二个轨道是上下两端的Li的1s轨道,能量和中间的Li的轨道很接近,但毕竟所处的化学环境有所不同,所以轨道能量也有点差别。上图第三个轨道是四个Li构成的4中心双电子轨道(可标注为4c-2e),显然是Li的2s电子构成的。由于本身单个原子的2s轨道能量就远高于1s,所以这个由2s轨道组成的LMO的能量也比其它两类LMO能量高一个数量级。
在前述的《使用AdNDP方法以及ELF/LOL、多中心键级研究多中心键》一文中我们用AdNDP方法也做过此体系的分析,得到的轨道图形和LMO几乎一模一样。因此,对于一些简单的体系,其实可以不必做操作相对麻烦的AdNDP,直接靠LMO就可以很好地讨论电子结构了。
我们再看看如果对此体系做NBO分析产生的轨道是什么样。我们用# b3lyp/6-311g(d) pop=saveNBO来计算,这样算完了之后NBO轨道就被直接存到chk里了,转换成fch后就可以直接用Multiwfn观看NBO轨道了,详见《使用Multiwfn绘制NBO及相关轨道》()。我们会发现能量最低的5个轨道,即Li的1s轨道,无论是轨道图形还是轨道能量与Multiwfn的轨道定域化产生的结果十分一致,但价层NBO轨道就一塌糊涂了。从输出信息中会看到除了CR型内核NBO轨道占据数几乎是2.0的,其余的占据数不太接近0的NBO的占据数为
6. (0.55080) LP*( 1)Li 1
7. (0.36965) LP*( 2)Li 1
10. (0.55080) LP*( 1)Li 2
11. (0.36965) LP*( 2)Li 2
14. (0.55080) LP*( 1)Li 3
15. (0.36965) LP*( 2)Li 3
18. (0.61749) LP ( 1)Li 4
22. (0.61749) LP ( 1)Li 5
可见,NBO对价层电子,没有给出任何成键信息,而假定每个原子上的价电子要么是孤对电子,要么是物理意义含糊不清的LP*电子。
(责任编辑:admin) |